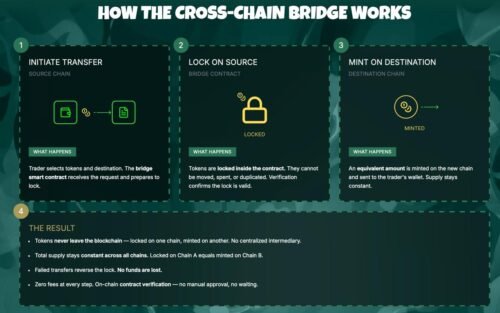
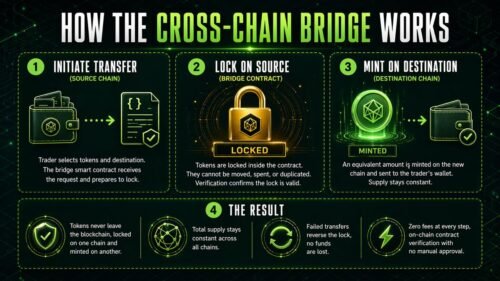
TORONTO, Ontario, Canada – July 18, 2026 – DapsyPay, a Canadian fintech company simplifying cross border payments, today announced the expansion of its international payment infrastructure to better support students, businesses, importers, and global entrepreneurs across Nigeria and other emerging markets.
As global commerce and international education continue to grow, millions of individuals and businesses still face slow settlements, expensive transfer fees, complex banking procedures, and payment limitations when sending money across borders. DapsyPay is addressing these challenges with a modern payment platform designed to make international transactions faster, more transparent, and easier to complete.
Headquartered in Canada, DapsyPay provides cross border payment solutions that enable users to pay international university tuition, settle supplier invoices, make business payments, and send funds globally through a streamlined digital platform.
“Our mission is simple: international payments should never be a barrier to education, business growth, or global opportunities,” said a spokesperson for DapsyPay. “We are building payment infrastructure that gives individuals and businesses a faster, more reliable way to move money across borders without the unnecessary complexity associated with traditional international transfers.”
Supporting International Students
International tuition payments remain one of the biggest challenges for students and parents studying abroad. Delayed bank transfers, poor exchange rates, payment limits, and lengthy processing times often create unnecessary stress during admission and enrollment.
DapsyPay is strengthening its payment capabilities to help students and families complete international tuition payments more efficiently, reducing delays while providing a smoother payment experience.
Empowering African Businesses
International payments are equally critical for businesses importing goods, paying overseas suppliers, settling invoices, and expanding into global markets.
DapsyPay is helping businesses across Nigeria and other African markets simplify cross border transactions with payment solutions built for today’s global economy.
By reducing friction in international payments, the company aims to help African businesses participate more confidently in international trade and unlock new opportunities for growth.
Building the Future of Cross Border Payments
The continued growth of digital commerce and international business has accelerated demand for faster, more flexible payment infrastructure.
DapsyPay plans to continue expanding its payment network, introducing additional international payment corridors, enhancing transaction capabilities, and investing in technologies that improve the speed, transparency, and accessibility of cross border payments.
As the company grows, its focus remains on delivering reliable international payment solutions for individuals, students, freelancers, importers, exporters, and businesses operating across multiple countries.
About DapsyPay
DapsyPay is a Canadian fintech company providing secure cross border payment solutions for individuals and businesses. The platform enables international tuition payments, supplier payments, business transfers, and other global transactions through a fast, transparent, and user friendly payment experience.
While headquartered in Canada, DapsyPay serves customers across Nigeria and other emerging markets, helping simplify international payments for education, commerce, and global business.
To learn more, visit https://dapsypay.com.
Media Contact
DapsyPay Media Relations
Email: support@dapsypay.com
Website: https://dapsypay.com